 9 minutes
9 minutes Ajouter un commentaire
Ajouter un commentaire
Les dystrophies rétiniennes héréditaires sont des maladies rares dégénératives de la rétine résultant de mutations de gènes cruciaux pour le développement et la fonction rétinienne (illustration).
LUXTURNA solution à diluer et solvant pour solution injectable (voretigène néparvovec) est
le premier traitement de thérapie génique de la dystrophie rétinienne héréditaire.
Il a obtenu une AMM européenne pour le traitement des patients adultes et des enfants présentant une perte visuelle due à une dystrophie rétinienne héréditaire résultant de mutations bi-alléliques confirmées du gène RPE65 et possédant suffisamment de cellules rétiniennes viables.
Il s'agit d'un médicament orphelin qui bénéficie en France d'une ATU (autorisation temporaire d'utilisation) de cohorte depuis octobre 2018.
 LUXTURNA fait l'objet d'une surveillance supplémentaire qui permettra l'identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de santé déclarent tout effet indésirable suspecté.
LUXTURNA fait l'objet d'une surveillance supplémentaire qui permettra l'identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de santé déclarent tout effet indésirable suspecté.Un vecteur viral associé à un gène pour corriger une mutation
Le mode d'action de LUXTURNA repose sur le transfert de matériel génétique dans les cellules rétiniennes grâce à un vecteur viral adéno-associé (AAV), le voretigène néparvovec. Ce dernier est dérivé d'un AAV présent naturellement et modifié en utilisant des techniques d'ADN recombinant.
Un mL de solution de LUXTURNA contient 5 x 1012 vg (génomes de vecteur) avant dilution.
L'injection de la solution de voretigène néparvovec dans l'espace sous-rétinien conduit à la transduction des cellules de l'épithélium pigmentaire rétinien par un ADNc codant la protéine RPE65 humaine normale, offrant le potentiel de restaurer le cycle visuel (Cf. Encadré 1).
Il semblerait que le voretigène néparvovec soit capturé par les cellules à travers les récepteurs protéoglycanes à héparine sulfate et dégradé par les voies cataboliques des protéines et de l'ADN endogènes.
Encadré 1 - Rôle du gène RPE65 dans les dystrophies héréditaires
| Les dystrophies rétiniennes héréditaires sont d'origine génétique ; elles se manifestent par une diminution de l'acuité visuelle et, à terme, une cécité. Parmi les dystrophies rétiniennes, on retrouve l'amaurose congénitale de Leber et la rétinite pigmentaire. Au cours de cette maladie génétique, le développement et la fonction rétinienne sont altérés à cause de mutations observées sur certains gènes cruciaux, dont le gène RPE65. Celui-ci est responsable de la production de la protéine RPE65 (retinal pigment epithelium 65 kDa), une protéine enzyme jouant un rôle majeur dans la restauration des photopigments actifs (bâtonnets et cônes). |
Une efficacité significativement supérieure à l'absence de traitement
Dans son avis du 3 avril 2019, la Commission de la Transparence a évalué l'efficacité et la tolérance de LUXTURNA sur la base principalement d'une étude de phase III comparative versus "absence de traitement", randomisée, ouverte, multicentrique (Lancet 2017 ; 390 : 849-86).
L'objectif de l'étude était de déterminer si l'administration séquentielle bilatérale sub-rétinienne du voretigène néparvovec améliore la capacité à se déplacer correctement dans l'espace (mesuré par un test de mobilité : Multi-Luminance Mobility Test) chez des enfants (> ou = 3 ans) et des adultes ayant une amaurose congénitale de Leber (ACL) résultant de mutations du gène RPE65.
Le critère de jugement principal était la variation du score du test de performance de mobilité en ambiance lumineuse variable (MLMT, Multi-Luminance Mobility Test) en vision binoculaire à 1 an par rapport à l'inclusion.
Au terme d'au moins 1 an de suivi, la variation moyenne du score MLMT (score de -1 à +6) a été significativement plus importante dans le groupe traité par voretigène néparvovec que dans le groupe sans traitement : +1,8 point versus +0,2 point respectivement, soit une différence moyenne de +1,6 point (IC95 % = [0,72 ; 2,41] ; p = 0,001).
Dans le groupe traité, 13/21 patients (61,9 %) ont été capables de réussir le test à 1 an avec une luminosité de 1 Lux, versus aucun patient dans le groupe non traité.
En termes de tolérance, les événements indésirables ont principalement été liés à la procédure d'administration avec, notamment, des risques potentiellement graves d'augmentation de la pression intra-oculaire, de déchirure rétinienne, de perforation maculaire, de maculopathie et d'inflammation oculaire, d'endophtalmie (1 cas rapporté au cours de l'étude).
Selon la Commission de la Transparence, un risque immunogène contre la capside virale et la protéine RPE65 doit être considéré à moyen et long terme bien que les données à court terme soient rassurantes.
SMR et ASMR importants
Sur la base des données disponibles, la Commission de la Transparence a attribué à LUXTURNA un service médical rendu (SMR) important et une amélioration du SMR également importante (ASMR II) dans la prise en charge des patients adultes et des enfants présentant une perte visuelle due à une dystrophie rétinienne héréditaire résultant de mutations bi-alléliques confirmées du gène RPE65 et possédant suffisamment de cellules rétiniennes viables.
Dans cette indication, la place de LUXTURNA est celle d'un traitement de 1re intention.
Si elle est favorable à l'agrément aux collectivités de LUXTURNA, la HAS soumet cette prise en charge à une réunion de concertation pluridisciplinaire pour décider de la mise en place de ce traitement. Cette décision doit reposer sur un faisceau d'examens, notamment pour déterminer le nombre de cellules rétiniennes viables suffisant, comportant un test génétique, des examens d'imagerie (tomographie en cohérence optique, optique adaptative), électrorétinogramme et des examens psychophysiques tels que la pupillométrie, l'acuité visuelle, le champ visuel et le test de mobilité en ambiance lumineuse variable (MLMT).
LUXTURNA en pratique
Le traitement doit être initié et administré par un chirurgien spécialiste de la rétine, expérimenté en chirurgie maculaire.
- Décongeler le flacon uniquement avant utilisation
Les flacons de LUXTURNA (produit et solvant) sont conservés au congélateur (température inférieure ou égale à - 65 °C), pendant 21 mois maximum.
Les flacons doivent être décongelés uniquement avant leur utilisation. La solution à injecter doit être préparée (dilution) dans les 4 heures précédant le début de la procédure d'administration, dans des conditions aseptiques (Cf. Monographie LUXTURNA - Rubrique Modalité Manipulation/Elimination).
Une fois décongelé, le médicament ne doit pas être recongelé et doit être laissé à température ambiante (moins de 25 °C). Après dilution, la solution doit être utilisée immédiatement ; si elle n'est pas utilisée immédiatement, le temps de stockage à température ambiante (moins de 25 °C) ne doit pas dépasser 4 heures.
- Injection au bloc opératoire
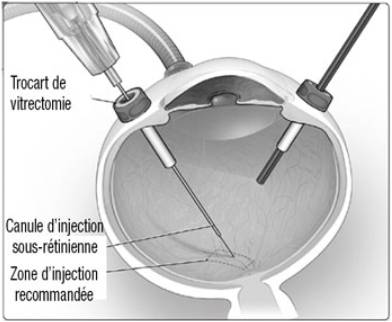
LUXTURNA est administré sous la forme d'une injection sous-rétinienne après vitrectomie dans chaque œil.
L'injection est réalisée au bloc opératoire en conditions aseptiques contrôlées.
Selon l'AMM, LUXTURNA peut être utilisé chez l'enfant à partir de 4 ans. En dessous de cet âge, la sécurité et l'efficacité du voretigène néparvovec n'ont pas été établies.
- Traitements complémentaires : immunomodulateurs et anesthésie
Tableau I - Traitement immunomodulateur pré- et post-opératoire pour chaque oeil
| Pré-opératoire | 3 jours avant l'administration de LUXTURNA | Prednisone (ou équivalent) 1 mg/kg/jour (maximum de 40 mg/jour) |
| Post-opératoire | 4 jours (incluant le jour d'administration) | Prednisone (ou équivalent) 1 mg/kg/jour (maximum de 40 mg/jour) |
| Suivi pendant 5 jours | Prednisone (ou équivalent) 0,5 mg/kg/jour (maximum de 20 mg/jour) | |
| Suivi pendant 5 jours d'une dose tous les deux jours | Prednisone (ou équivalent) 0,5 mg/kg un jour sur deux (maximum de 20 mg/jour) |
- Ecarter toute infection avant d'administrer LUXTURNA et surveiller la pression oculaire
La pression intra-oculaire doit être surveillée avant et après l'administration du médicament, afin qu'elle soit prise en charge de manière appropriée.
- Précautions de manipulation
Ces précautions de manipulation doivent être suivies pendant 14 jours après l'administration de voretigène néparvovec.
Il est recommandé aux patients et au personnel soignant de porter des gants pour le changement des pansements et l'élimination des déchets, particulièrement dans les cas de grossesse, d'allaitement et d'immunodéficience du personnel soignant.
Conseils aux patients : effets indésirables, voyages et activité physique
Les patients doivent être prévenus de la survenue de troubles visuels temporaires, comme une vision trouble et une photophobie, durant les semaines suivant le traitement.
En cas de persistance de ces signes, une consultation médicale est nécessaire.
Les patients ne doivent pas se baigner en raison d'un risque accru d'infection oculaire, et éviter une activité physique intense en raison d'un risque accru de lésion oculaire. La baignade et l'activité intense sont à nouveau autorisés après au moins une à deux semaines, sur les conseils du médecin.
Du fait d'un risque d'augmentation de la pression intra-oculaire, les voyages en avion ou autres voyages EN altitude élevée sont à éviter jusqu'à ce que la bulle d'air formée lors de l'administration de LUXTURNA se soit totalement dissipée.
Une période d'une semaine ou plus après l'injection peut être nécessaire avant la dissipation de la bulle d'air qui doit être vérifiée à l'examen ophtalmique. Une augmentation rapide de l'altitude alors que la bulle d'air est toujours présente peut entraîner une augmentation de la pression intra-oculaire et une perte irréversible de la vision.
Identité administrative
- Réservé à l'usage hospitalier
- Prescription réservée aux spécialistes en ophtalmologie
- Boîte de 1 flacon de 2 mL + 2 flacons de solvant, CIP 3400955060796
- Prise en charge par les collectivités en relais de l'ATU de cohorte (demande d'inscription d'agrément aux collectivités en cours)
- Laboratoire Novartis Pharma
La prise en charge est limitée au traitement des patients adultes et des enfants présentant une perte visuelle due à une dystrophie rétinienne héréditaire résultant de mutations bi-alléliques confirmées du gène RPE65 et possédant suffisamment de cellules rétiniennes viables.
Elle est subordonnée aux conditions de prescription et au mode d'organisation des soins suivants :
- la décision de mise sous traitement doit faire l'objet d'une réunion de concertation pluridisciplinaire et doit reposer sur un faisceau d'examens, notamment pour déterminer le nombre de cellules rétiniennes viables suffisant, comportant un test génétique, des examens d'imagerie (tomographie en cohérence optique, optique adaptative), électrorétinogramme et des examens psychophysiques tels que la pupillométrie, l'acuité visuelle, le champ visuel et le test de mobilité en ambiance lumineuse variable (MLMT).
Pour aller plus loin
Avis de la Commission de la Transparence - LUXTURNA (HAS, 3 avril 2019)
Etude pivot
Russell S, Bennett J, Wellman JA et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy : a randomised, controlled, open-label, phase 3 trial. Lancet. 2017 ; 390 : 849-860
Pour aller plus loin
Consultez les monographies VIDAL
Sources
Pour recevoir gratuitement toute l’actualité par mail Je m'abonne !


Commentaires
Cliquez ici pour revenir à l'accueil.