 8 minutes
8 minutes Ajouter un commentaire
Ajouter un commentaire
Représentation en 3D d’un lymphocyte T activé dans le cadre d’une immunothérapie (illustration).
TECENTRIQ 1 200 mg solution à diluer pour perfusion (atézolizumab) est un antinéoplasique hospitalier indiqué dans les situations suivantes :
- en monothérapie, dans le traitement des patients adultes atteints d'un cancer bronchique non à petites cellules (CBNPC) localement avancé ou métastatique après une chimiothérapie antérieure (Cf. VIDAL Reco "Cancer du poumon"). Les patients avec mutations activatrices de l'EGFR ou réarrangement du gène ALK (ALK-positif) doivent également avoir reçu une thérapie ciblée avant de recevoir TECENTRIQ ;
- en monothérapie, dans le traitement des patients adultes atteints d'un carcinome urothélial localement avancé ou métastatique :
- après une chimiothérapie antérieure à base de platine,
- ou considérés inéligibles au cisplatine et dont les tumeurs présentent une expression de PD-L1 supérieure ou égale à 5 %.
Le 5 avril 2019, l'ANSM (Agence nationale de santé du médicament et des produits de santé) a octroyé à TECENTRIQ une ATU (autorisation temporaire d'utilisation) de cohorte d'extension d'indication en association au carboplatine et à l'étoposide, en première ligne de traitement des patients adultes atteints d'un cancer bronchique à petites cellules (CBPC) de stade étendu et présentant un score ECOG de 0 ou 1.
Cette extension d'indication est encadrée par un protocole d'utilisation thérapeutique et de recueil d'information, validé le 3 mai 2019.
 TECENTRIQ fait l'objet d'une surveillance supplémentaire qui permettra l'identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté.
TECENTRIQ fait l'objet d'une surveillance supplémentaire qui permettra l'identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté.NB. Le 5 mars 2019, la Commission Européenne a adopté l'extension d'indication de TECENTRIQ "en association au bévacizumab, paclitaxel et carboplatine, en première ligne de traitement du cancer bronchique non à petites cellules (CBNPC) non épidermoïde métastatique. Chez les patients atteints d'un CBNPC avec EGFR muté ou réarrangement du gène ALK (ALK-positif), TECENTRIQ, en association au bévacizumab, paclitaxel et carboplatine, est indiqué seulement après échec des thérapies ciblées appropriées".
L'atézolizumab, un anticorps monoclonal anti-PD-L1
Le principe actif de TECENTRIQ, l'atézolizumab, est un anticorps monoclonal humanisé de type immunoglobuline G1 (IgG1) anti-PD-L1 (programmed death-ligand 1, ligand de mort programmée 1).
La protéine PD-L1 peut être exprimée sur les cellules tumorales et/ou sur les cellules immunitaires infiltrant la tumeur et contribuer à l'inhibition de la réponse immunitaire anti-tumorale dans le micro-environnement tumoral.
La liaison de PD-L1 aux récepteurs PD-1 et B7.1, présents sur les cellules T et sur les cellules présentatrices d'antigène, inhibe l'activité cytotoxique des cellules T, la prolifération des cellules T et la production de cytokines.
En se liant directement à PD-L1, l'atézolizumab assure un double blocage des récepteurs PD-1 et B7.1, empêchant ainsi l'inhibition de la réponse immunitaire médiée par PD-L1/PD-1 et réactivant la réponse immunitaire antitumorale, sans induire de cytotoxicité cellulaire anticorps-dépendante (Cf. Figure 1).
Figure 1 - Mécanisme d'action de l'atézolizumab
(d'après le dossier de presse TECENTRIQ du laboratoire Roche)
(d'après le dossier de presse TECENTRIQ du laboratoire Roche)
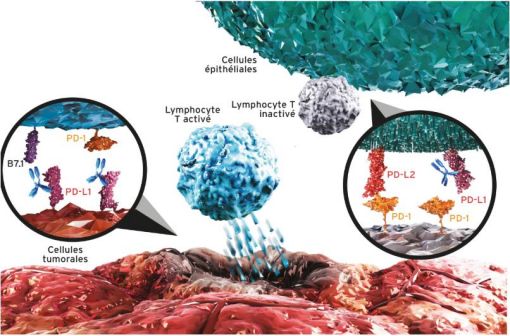
L'atézolizumab n'affecte pas l'interaction PD-L2/PD-1, impliquée dans l'homéostasie du système immunitaire.
L'atézolizumab est le premier anti-PD-L1 bénéficiant d'une AMM (autorisation de mise sur le marché) dans le cancer bronchique non à petites cellules.
L'atézolizumab supérieur au docétaxel en termes de médiane de survie globale dans le CBNPC (étude OAK)
Dans son avis du 30 mai 2018, la Commission de la Transparence a évalué l'efficacité et la tolérance de TECENTRIQ dans le CBNPC sur la base principalement de l'étude pivot de phase III OAK (Lancet 2017), ouverte, randomisée, comparative versus docétaxel (1 200 mg d'atézolizumab toutes les 3 semaines versus 75 mg/m² toutes les 3 semaines).
Les patients inclus (n = 850) étaient atteints d'un CBNPC et avaient reçu un traitement par chimiothérapie à base de sels de platine. Dans cette cohorte, 10 % des patients avaient une mutation connue de l'EGFR (Epidermal Growth Factor Receptor) et 0,2 % (n = 2) des réarrangements du gène ALK (Anaplastic lymphoma kinase).
Le critère de jugement principal était la survie globale définie comme le délai entre le jour de la randomisation et la date de décès du patient, toutes causes confondues, évaluée dans deux co-populations :
- population primaire (n = 850)
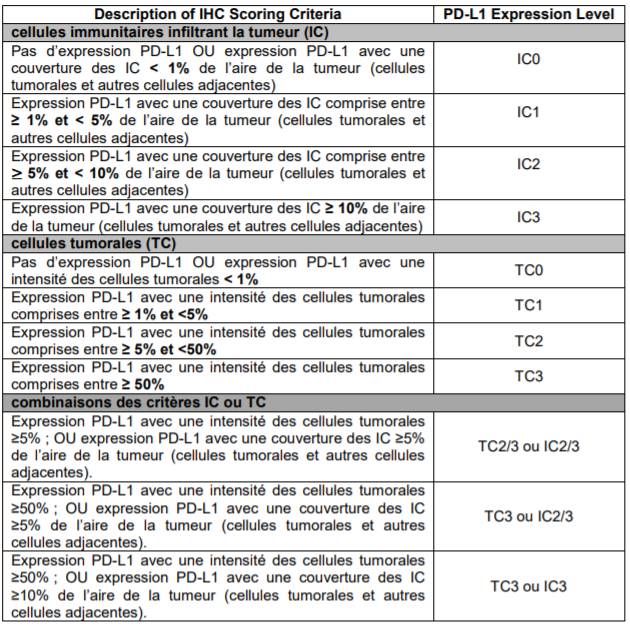
- sous-groupe IC1/2/3 ou TC1/2/3 (PD-L1 > ou = 1%) [Cf. Tableau I].
Tableau I - Critères pour l'évaluation de l'expression PD-L1
Selon les résultats de cette étude à la date du 7 juillet 2016 et avec un suivi médian d'environ 21 mois, la supériorité de l'atézolizumab par rapport au docétaxel a été démontrée dans les deux populations étudiées avec :
- dans la population constituée des 850 premiers patients randomisés (parmi les 1 225 prévus) : 13,8 mois de médiane de survie globale dans le groupe atézolizumab versus 9,6 mois dans le groupe docétaxel, soit un gain en valeur absolue de 4,2 mois (HR = 0,73 IC95 % [0,62 ; 0,87] ; logrank stratifié p = 0,0003, inférieur au seuil fixé à 3 %) ;
- dans le sous-groupe de patients ayant une expression PD-L1 IC1/2/3 ou TC1/2/3 (n = 463 patients parmi les 850 premiers patients randomisés) : 15,7 mois de médiane de survie globale dans le groupe atézolizumab versus 10,3 mois dans le groupe docétaxel, soit un gain en valeur absolue de 5,4 mois (HR = 0,74 IC95 % [0,58 ; 0,93] ; logrank stratifié p = 0,0102, inférieur au seuil fixé à 2 %).
En termes de tolérance, les données de l'étude OAK indiquent que les événements indésirables ont été plus fréquents dans le groupe docétaxel que dans le groupe atézolizumab, notamment les événements indésirables conduisant à l'arrêt du traitement (18,7 % versus 7,6 %) et les événements indésirables de grades 3-4 (53,6 % versus 37,3 %).
A l'inverse, les événements indésirables d'intérêt particulier d'ordre dermatologique (14,4 % versus 10,4 %), hépatique (8,2 % versus 2,6 %) ou endocrinologique (5,6 % versus 0,3 %) ont été plus fréquents dans le groupe atézolizumab par rapport au docétaxel.
S'agissant des risques importants identifiés dans le plan de gestion des risques (PGR), ils concernent les réactions liées à la perfusion et les effets indésirables d'origine immunologique : pneumopathie inflammatoire, colite, hépatite, pancréatite, endocrinopathie (incluant maladie inflammatoire de l'hypophyse, de la thyroïde et diabète), méningo-encéphalite, neuropathie (incluant le syndrome de Guillain Barré et le syndrome myasthénique).
Des données de tolérance à long terme ne sont pas disponibles.
SMR important dans le CBNPC sur un périmètre restreint par rapport à l'AMM
Tenant compte de l'ensemble des données disponibles lors de son évaluation, la Commission de la Transparence considère qu'après échec d'une chimiothérapie à base de sels de platine, en 2e ligne ou plus, TECENTRIQ en monothérapie représente une alternative au nivolumab (OPDIVO) ou au pembrolizumab (KEYTRUDA, uniquement en cas de tumeur avec surexpression PD-L1 > ou = 1 %) chez les patients ayant un CBNPC localement avancé ou métastatique, épidermoïde ou non épidermoïde.
Chez les patients EGFR mutés, la place de l'atézolizumab reste à étudier avec un niveau de preuve optimal. Chez les patients ayant un réarrangement ALK (ALK+), l'atézolizumab n'a pas de place dans la stratégie thérapeutique faute de données cliniques.
En conclusion, la Commission considère que le service médical rendu (SMR) par TECENTRIQ en monothérapie est :
- important dans le traitement des patients adultes atteints d'un CBNPC localement avancé ou métastatique après une chimiothérapie antérieure, les patients avec mutations activatrices de l'EGFR devant également avoir reçu une thérapie ciblée,
- insuffisant dans le traitement des patients adultes atteints d'un CBNPC localement avancé ou métastatique, avec réarrangement du gène ALK (ALK+), après une chimiothérapie antérieure.
Dans le traitement du carcinome urothélial, TECENTRIQ n'a pas été évalué par la HAS (Haute Autorité de Santé) du fait que le laboratoire n'a pas demandé l'inscription au remboursement dans cette indication.
Sécurité des patients : mesures de réduction des risques
En complément de l'AMM (autorisation de mise sur le marché), des mesures additionnelles sont mises en place auprès des professionnels de santé et des patients pour réduire les risques associés à TECENTRIQ.
Ces mesures comportent :
- pour les médecins : le résumé des caractéristiques du produit (RCP) de TECENTRIQ et une brochure d'information ;
- une carte alerte patient remise par le prescripteur, mentionnant les messages clés et notamment les signes et symptômes évoquant la survenue d'un effet indésirable. Le patient doit conserver cette carte sur lui en permanence.
TECENTRIQ en pratique
La dose recommandée de TECENTRIQ est de 1 200 mg administrée par voie intraveineuse toutes les 3 semaines, quelle que soit son indication.
TECENTRIQ est administré par voie IV (intraveineuse), en perfusion de 60 minutes.
La solution (20 mL) doit être diluée dans une poche de perfusion (250 mL), avec du chlorure de sodium 0,9 %. Après la dilution, 1 mL de solution contient environ 4,4 mg de TECENTRIQ.
Si la première perfusion est bien tolérée, toutes les perfusions suivantes peuvent être administrées en 30 minutes.
En raison de ses effets indésirables d'origine immunologique ou liés à la perfusion, les patients traités par TECENTRIQ nécessitent une surveillance particulière (Cf. Tableau II) en vue d'une identification rapide et d'une prise en charge dans les meilleurs délais.
Tableau II - Surveillance biologique des patients sous TECENTRIQ
| Toxicité hépatique | Dosage de l'aspartate aminotransférase (ASAT), l'alanine aminotransférase (ALAT) et de la bilirubine avant et pendant le traitement. |
| Toxicité endocrinienne | Surveillance de la fonction thyroïdienne avant et pendant le traitement. |
| Toxicité pancréatique | Surveillance du taux sérique d'amylacé ou de lipase. |
Le traitement doit être poursuivi jusqu'à perte du bénéfice clinique ou survenue d'une toxicité inacceptable.
En présence d'effets indésirables et selon leur sévérité, le traitement doit être suspendu ou arrêté définitivement.
Les patients traités par TECENTRIQ doivent recevoir la carte alerte patient et être informés des risques liés à l'utilisation de ce traitement.
Identité administrative
- Liste I
- Réservé à l'usage hospitalier
- Prescription réservée aux spécialistes et services de cancérologie et d'oncologie médicale
- Surveillance particulière pendant le traitement
- Flacon de 20 mL, boîte unitaire, CIP 3400955042006, UCD 3400894312628
- Agrément aux collectivités - Cf. Encadré ci-après (Journal officiel du 20 février 2019 - texte 9)
- Inscription en sus des prestations d'hospitalisation (Journal officiel du 20 février 2019 - texte 7)
- Laboratoire Roche
Périmètre de prise en charge
| Traitement des patients adultes atteints d'un cancer bronchique non à petites cellules (CBNPC) localement avancé ou métastatique après une chimiothérapie antérieure, les patients avec mutations activatrices de l'EGFR devant également avoir reçu une thérapie ciblée. |
Pour aller plus loin
Avis de la Commission de la Transparence - TECENTRIQ et cancer pulmonaire (HAS, 30 mai 2018)
L'ANSM octroie une ATU de cohorte d'extension d'indication pour l'utilisation de Tecentriq (atezolizumab) dans le cancer bronchique à petites cellules - Point d'Information (6 mai 2019)
Carte Alerte Patient
Brochure d'information pour les professionnels de santé
Etude pivot :
Rittmeyer A., Barlesi F. Waterkamp D. et al. Atezolizumab versus docetaxel in patients with previously treated nonsmall-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. The Lancet 2017, 389 : 255- 265 (abstract)
Sur VIDAL.fr
KEYTRUDA (pembrolizumab) et TECENTRIQ (atezolizumab) : restriction d'indication dans la prise en charge du carcinome urothélial (12 juillet 2018)
Pour aller plus loin
Consultez les monographies VIDAL
Sources
Pour recevoir gratuitement toute l’actualité par mail Je m'abonne !


Commentaires
Cliquez ici pour revenir à l'accueil.