 7 minutes
7 minutes Ajouter un commentaire
Ajouter un commentaire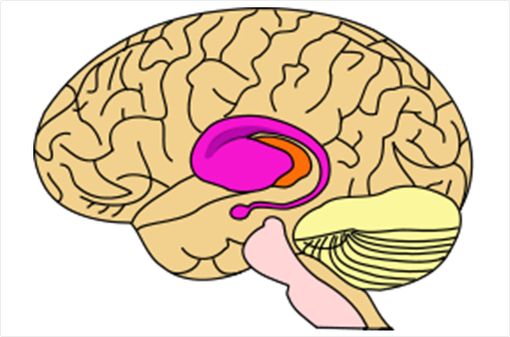
La maladie de Huntington est une affection neurodégénérative rare qui affecte surtout le striatum, composé du noyau caudé et du putamen (partie rose du schéma ci-dessus, © Wikipedia).
La maladie de Huntington, une affection d'origine génétique
La maladie de Huntington est une affection neuro-dégénérative rare (5 à 10 personnes touchées pour 100 000), liée à la mutation d'un gène du chromosome 4 (mutation découverte par MacDonald et coll., Cell 1993). Ce gène, appelé 4p16.3, code normalement pour une protéine, la Huntingtine, qui régule plusieurs fonctions cellulaires.
Lorsque ce gène est muté, il comporte davantage de triplets de nucléotides CAG (cytosine, adénine et guanine), ce qui altère la Huntingtine et déclencherait la maladie (comme le souligne la HAS, le mécanisme physiopathologique de cette pathologie n'est pas encore complètement élucidé).
Les lésions des neurones consécutives à ce dysfonctionnement protéique sont situées surtout au niveau du striatum (structure composée du noyau caudé et du putamen, située au milieu du cerveau).
Le plus souvent, cette maladie se déclenche entre 30 et 50 ans, mais il existe des formes juvéniles, qui débutent avant l'âge de 20 ans, ainsi que des formes plus tardives.
La transmission de cette mutation est autosomique dominante : une personne non atteinte ne peut pas transmettre la maladie ; une personne atteinte a 50 % de risques de la transmettre à chacun de ses enfants.
Un diagnostic avant tout clinique
Georges Huntington, médecin généraliste américain, a décrit pour la première fois la pathologie qui portera ensuite son nom lors d'un discours prononcé en 1872. Il explique avoir constaté l'existence d'une chorée (cf. infra) héréditaire, associée à "une tendance à la folie" et apparaissant vers 40 ans.
De fait, le tableau clinique, lié à la lésion puis destruction des neurones, peut associer :
- des mouvements typiques appelés "chorée", ou "syndrome choréique" : il s'agit d'une succession de mouvements "anormaux, involontaires, incontrôlables, brusques, explosifs, anarchiques, imprévisibles, irréguliers, de courte durée, non rythmiques", résume le PNDS. Ces mouvements choréiques, présents chez environ 90 % des patients atteints, sont augmentés par les émotions, le stress et la concentration, et disparaissent le plus souvent pendant le sommeil ;
- D'autres troubles moteurs : Les patients atteints peuvent présenter des dystonies (troubles du tonus musculaire entraînant des postures anormales), une rigidité (pouvant poser un problème de diagnostic différentiel avec la maladie de Parkinson), une dysphagie (troubles de la déglutition), des myoclonies (contractions musculaires brusques, brèves et involontaires), des troubles de la marche et de l'équilibre (apparition progressive, en rapport avec l'intensité des autres symptômes moteurs), un bruxisme (serrement involontaire des dents) ou encore une perturbation de la dextérité manuelle ;
- des troubles cognitifs : troubles des fonctions exécutives (attention, capacité d'initiation, flexibilité, planification), bradyphrénie (ralentissement du traitement cognitif de l'information, allongement du temps de réaction), troubles du langage (articulation ou émission de sons défaillante, réduction de la fluence verbale, simplification syntaxique, etc.), troubles de la relation à autrui (troubles du comportement, changements de personnalité), baisse de la mémoire (troubles mnésiques), désorientation, troubles visuo-spatiaux et visuo-perceptifs ;
- des troubles psychiatriques : dépression, suicide (risque suicidaire élevé, corrélé aux antécédents dépressifs), irritabilité et/ou agressivité (30 à 65 % des patients), apathie, anxiété, obsessions, impulsivité, impatiences (akathisie : impossibilité de rester en place), troubles du comportement sexuel, hallucinations ou idées délirantes, agitation ;
- des troubles somatiques : troubles du sommeil, incontinence, pathologies dentaires, troubles digestifs, transpiration excessive (hyperhydrose), douleur, perte de poids (souvent précoce, parfois antérieure à l'apparition des autres symptômes), hypersalivation (sialorrhée).
L'association de mouvements choréiques, de troubles cognitifs et/ou psychiatriques peut faire évoquer ce diagnostic, confirmé par l'histoire familiale et/ou la mise en évidence de la mutation du chromosome 4.
Une prise en charge thérapeutique symptomatique et pluridisciplinaire
Il n'existe malheureusement pas, pour le moment, de traitement curatif de la maladie de Huntington (autrefois appelée "chorée de Huntington", mais ce terme réduisait cette affection à ses symptômes moteurs). Les troubles moteurs, cognitifs et psychiatriques s'aggravent progressivement, conduisant en une vingtaine d'années, en moyenne, à une dégradation globale.
Par contre, cette évolution péjorative peut être endiguée, ralentie par une prise en charge symptomatique pluridisciplinaire : médecin traitant, médecin du centre de référence, spécialistes (neurologue, généticien, psychiatre, médecin de rééducation, urgentistes, etc.), professionnels paramédicaux (infirmier, masseur-kinésithérapeute, psychologue, psychomotricien, orthophoniste, diététicien, assistante sociale, etc.).
L'objectif de cette prise en charge "est de préserver le plus longtemps possible l'autonomie du patient, la vie professionnelle et sociale, la qualité de vie, l'harmonie familiale et les ressources financières", résume le PNDS de la HAS.
Exemples de traitements symptomatiques recommandés
Le PNDS recommande des traitements symptomatiques, médicamenteux ou non, pour chacun des symptômes évoqués ci-dessus.
Voici des exemples des attitudes thérapeutiques préconisées par ce protocole :
- syndrome choréique : la tétrabénazine a montré un effet bénéfique dès le début de traitement (grade de cette recommandation selon l'échelle établie par la HAS : Grade A). Les neuroleptiques atypiques, "bien que n'ayant pas l'AMM dans cette indication, constituent le premier choix de traitement lorsque le patient présente en plus de la chorée, des troubles du caractère, du comportement, une dépression active ou des troubles psychotiques" (Grade C). En seconde intention, d'autres neuroleptiques ont une AMM (autorisation de mise sur le marché) pour la chorée (halopéridol, tiapride et pimozide) (Accord Pro). La monothérapie "doit être préférée". Des mesures non médicamenteuses peuvent aussi s'avérer très utiles : mesures de protection pour éviter au patient de se blesser (aménagement de son intérieur), relaxation, yoga, balnéothérapie, tai chi chuan, qi gong, gymnastique douce, danse, rééducation, sports non vulnérants comme la marche (Accord Pro) ;
- dystonie : rééducation, benzodiazépines (en particulier le clonazepam), voire toxine botulique (dystonie focale) (Accord Pro);
- rigidité : la lévodopa peut permettre une amélioration, partielle et transitoire (Grade C). Les agonistes dopaminergiques et la mantadine peuvent être utiles, en particulier en cas d'échec de la lévodopa (Grade C) ;
- myoclonies : en cas de myoclonies d'origine corticale (cérébrale) plus ou moins associées à des crises d'épilepsie cliniques, le PNDS "recommande l'essai du valproate de sodium ou du clonazepam, en monothérapie ou en association, à des doses progressives" (Grade C). Le levetiracetam est une alternative thérapeutique dans les mêmes indications (Accord pro) ;
- troubles de la marche et de l'équilibre : rééducation, aides techniques adaptées (déambulateur à 4 roues, également appelé "rollator", canne, fauteuil roulant) et/ou port de protections (Accord pro). Les traitements médicamenteux utilisés pour réduire le syndrome choreique peuvent améliorer ces troubles (Grade C) ;
- troubles cognitifs : aide à l'élaboration des tâches et activités du quotidien, à la mise en place de routines, de repères, et aide aux aidants (entourage familial, auxiliaire de vie, aide éducative, etc.) (Accord pro) ;
- troubles psychiatriques : traitement d'une dépression (prise en charge psychologique Accord Pro, inhibiteur sélectif de la sérotonine Grade C, traitement thymorégulateur si besoin Accord pro, électroconvulsivothérapie "en cas de dépression sévère résistant aux médicaments per os, plus ou moins associée à des éléments psychotiques" Grade C), d'une agressivité excessive (stratégies comportementales Accord pro, éventuellement neuroleptiques atypiques utilisés hors AMM comme l'olanzapine ou la rispéridone Grade C), d'autres symptômes psychiatriques en fonction de leur gravité (et des médicaments pris pour les autres symptômes) ;
- troubles somatiques : règles hygiéno-diététiques pour améliorer le sommeil, les troubles digestifs ou encore la perte de poids, traitement d'une insomnie persistante (hypnotique de courte durée, voire neuroleptique), d'une incontinence urinaire (la carbamazépine peut être efficace dans certains cas Grade C, ou d'antimuscariniques comme le trospium ou la solifénacine Accord pro), d'une hypersalivation (scopolamine percutanée, atropine per os ou autres anticholinergiques, "en se méfiant du risque iatrogène, en particulier syndrome confusionnel, constipation et rétention urinaire" Accord pro ; des injections de toxine botulique dans les glandes salivaires peuvent être envisagées Accord pro).
L'éducation thérapeutique (information, explications, apprentissage, accompagnement) et le recours aux associations de patients (cf. liens ci-dessous) peuvent aider le patient et son entourage à faire face et vivre avec cette terrible maladie...
En conclusion
La maladie de Huntington, qui associe des troubles progressivement invalidants, peut être moins pénible au quotidien et son évolution relativement ralentie avec une prise en charge multidisciplinaire ajustée aux particularités de chaque patient. Les détails de cette prise en charge, brièvement résumée ci-dessus, sont présents dans le PNDS publié par la HAS.
Du côté de la recherche, comme pour d'autres maladies neurodégénératives, les scientifiques travaillent sur des pistes de greffes de neurones fœtaux ou "souches" (dans la zone cérébrale affectée, le striatum) et de thérapie génique (agir sur la protéine Huntingtine pour qu'elle ne soit plus altérée, ou inhibition de sa production, ou correction du gène muté). Mais pour l'instant, aucun de ces traitements n'est malheureusement encore utilisable.
Pour en savoir plus :
Protocole national de diagnostic et de soins pour les maladies rares : Prise en charge médicamenteuse de la maladie de Huntington, HAS, août 2015
A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes, MacDonald ME et coll., Cell, mars 1993
"On Chorea" by George Huntington, M.D., The Medical and Surgical Reporter: A Weekly Journal, (Philadelphia: S.W. Butler), vol. 26, no. 15 (April 13, 1872), pp. 317-321.
Site du Centre de référence National pour la maladie de Huntington, coordonné par le Pr Bachoud Lévi, CHU Henri Mondor, Créteil
La maladie de Huntington sur Orphanet, le portail des maladies rares et médicaments orpheins ; Fiche explicative en format PDF.
La maladie de Huntington sur le site du Centre de référence maladies neurogénétiques du CHU d'Angers
L'association Huntington France
L'association Huntington Avenir
L'association DingDingDong
Sur VIDAL.fr :
VIDAL Reco Maladies rares : maladie de Huntington
Sources
Pour recevoir gratuitement toute l’actualité par mail Je m'abonne !
.jpg)

Commentaires
Cliquez ici pour revenir à l'accueil.